Científicos y científicas lograron secuenciar más de 400 genomas virales de SARS-CoV2 en pacientes de todo el país
El Consorcio, que realiza estudios genómicos de SARS-CoV2 en nuestro país para aportar a los datos globales de circulación viral en la base de datos GISAID, brinda información precisa de los distintos linajes del virus y como estos circulan en nuestra población.
Creado desde el Ministerio de Ciencia, Tecnología e Innovación de la Nación, el "Consorcio interinstitucional para la Secuenciación del genoma y estudios genómicos de SARS-CoV2 (Proyecto PAIS)", conformado por grupos de investigación de diferentes instituciones con el objetivo de secuenciar el genoma y realizar demás estudios genómicos del SARS-CoV2, logró secuenciar más de 400 nuevos genomas de SARS-CoV2 en Argentina.
Coordinado por la Dra. Mariana Viegas del Laboratorio de Virología Hospital de Niños Ricardo Gutiérrez, el equipo trabaja articulada y colaborativamente para realizar estudios genómicos de SARS-CoV2 en nuestro país y aportar tanto al conocimiento local como a la base de datos global de circulación viral GISAID (Global Initiative on Sharing All Influenza Data). Entre sus objetivos se encuentran la secuenciación de los genomas circulantes de SARS-CoV2 en distintas regiones de nuestro país, el análisis a gran escala de secuencias, ensamblado de genomas, análisis filogenéticos y filogeográficos, epidemiología y evolución molecular, y estudios de correlación clínica.
En su más reciente estudio, el Consorcio ha obtenido 305 nuevos genomas de SARS-CoV2, 282 provenientes del AMBA (CABA y GBA) y 23 de otras zonas de la Provincia de Buenos Aires. Del mismo, participaron tres de los siete nodos de secuenciación que forman parte del consorcio: Laboratorio de Virología Hospital de Niños Ricardo Gutiérrez (CABA), Biocódices (San Martín, provincia de Buenos Aires) y IABIMO-INTA Castelar (Hurlingham, provincia de Buenos Aires). Asimismo, han participado 15 centros de Salud distribuidos entre CABA y provincia de Buenos Aires que realizan el diagnóstico diario de COVID-19 y que aportaron muestras y datos clínico-epidemiológicos de los casos secuenciados. El análisis de los datos estuvo a cargo de los nodos de bioinformática y evolución que integran el Consorcio.
AMBA:
Se seleccionaron muestras tomadas entre el 28 de marzo y el 12 de junio. En este período, los datos epidemiológicos mostraron un aumento de circulación comunitaria de SARS-CoV-2 en el AMBA. El objetivo de este nuevo ciclo de secuenciación fue identificar el establecimiento de grupos de virus de transmisión locales y su distribución en las regiones abarcadas.
Los resultados principales del trabajo son los siguientes:
-
Algunos de los genomas presentaron cambios que podrían impactar en sus características biológicas o su diagnóstico molecular. Estos hallazgos de vigilancia epidemiológica molecular serán estudiados para establecer su alcance e implicancia.
-
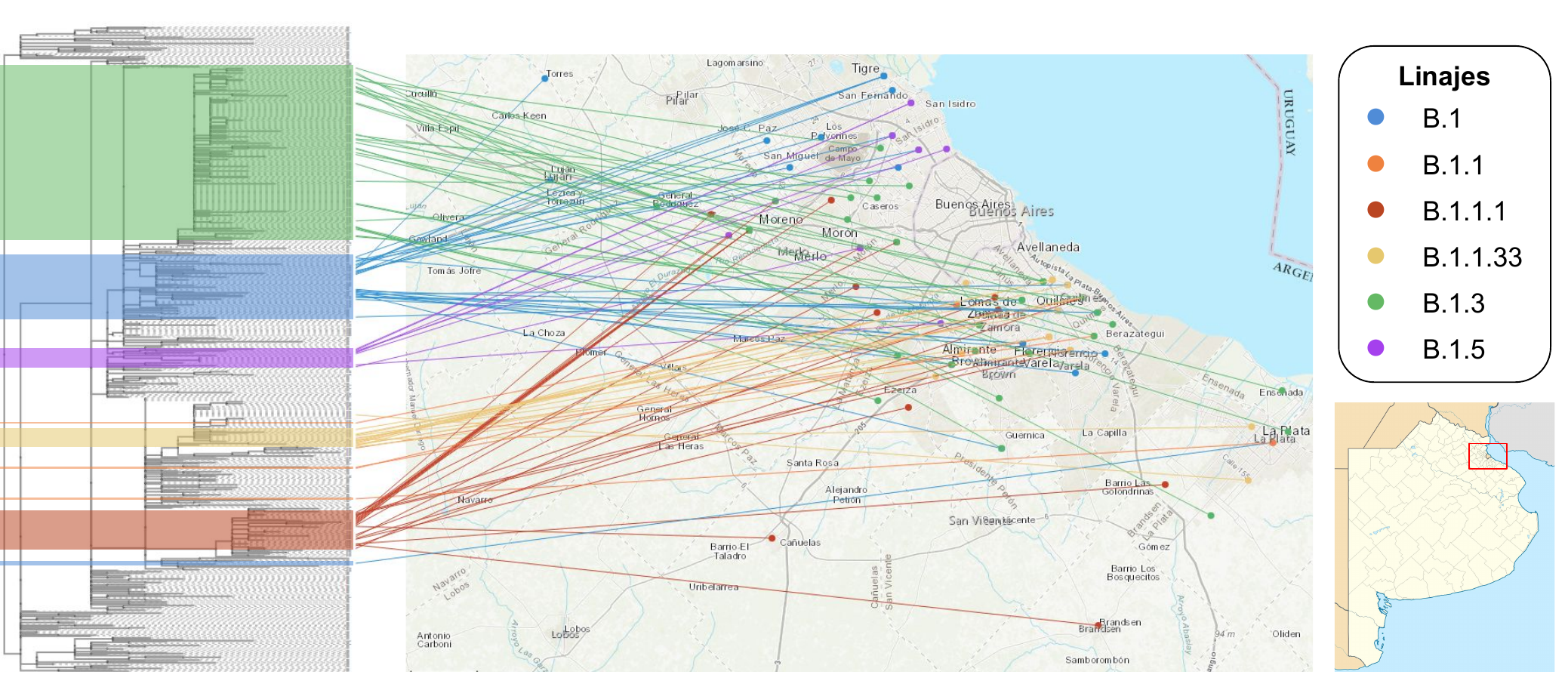
La distribución de los linajes virales de las 122 muestras de la provincia de Buenos Aires fue la siguiente: B.1.3 (32,8 %), B.1 (23,0 %), B.1.1.1 (19,7 %), B.1.1.33 (13,9 %), B.1.5 (6,6 %) y B.1.1 (4,1 %).
-
Se observó una marcada regionalización de los clusters genéticos, muchos de estos compatibles con circulación viral comunitaria sostenida restringida a zonas geográficas específicas (GBA-Norte, GBA-Oeste y GBA-Sur). Si bien en algunos casos se observó relación cercana entre secuencias de GBA y secuencias de CABA, este patrón no fue el predominante.
-
Los análisis realizados mostraron evidencias de distintas introducciones de virus y su diversificación en zonas circunscritas, más que la circulación irrestricta entre las distintas zonas estudiadas. Este proceso de diversificación viral es compatible con las restricciones de circulación poblacional debido al Aislamiento Social Preventivo y Obligatorio.
-
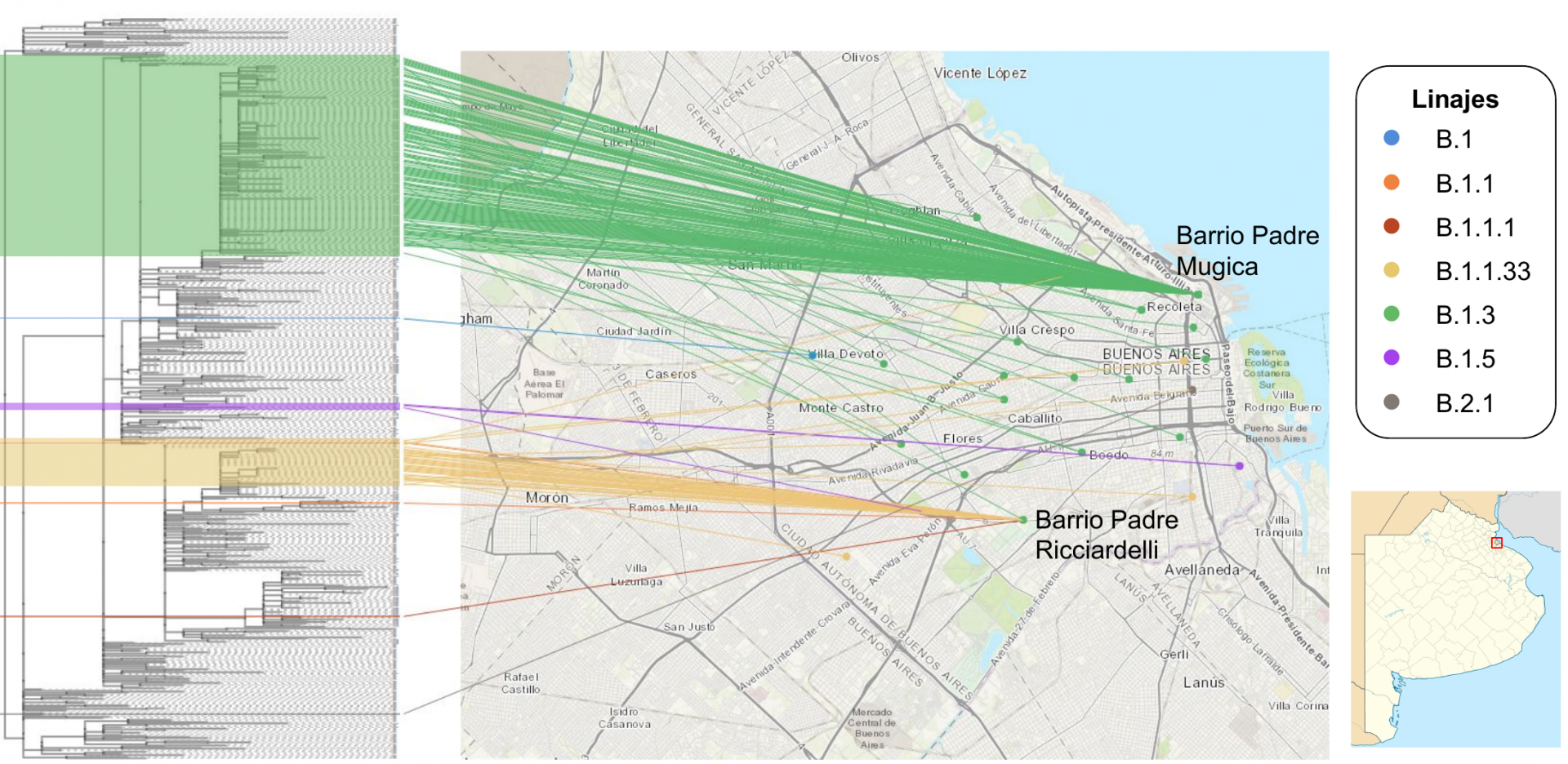
La distribución de los linajes virales de las 183 muestras de la Ciudad Autónoma de Buenos Aires fue la siguiente: B.1.3 (76,0 %), B.1.1.33 (19,1 %), B.1.5 (1,6 %), B.1 (1,1 %), B.1.1 (0,5 %), B.1.1.1 (0,5 %), y linaje B.2 (0,5 %) y B.2.1 (0,5 %).
-
Un muestreo intensivo en los Barrios populares Padre Mugica y Padre Ricciardelli mostró una gran homogeneidad en las variantes virales, compatible con una alta transmisibilidad en zonas de conglomerado. Sin embargo, las variantes responsables de los brotes en cada uno de los barrios fueron diferentes (B.1.3 para el primero y B.1.1.33 para el segundo). La presencia de secuencias de SARS-CoV-2 muestreadas en el mes de abril y previamente reportadas por el Consorcio indicarían que tanto el linaje B.1.3 como el B.1.1.33 estuvieron presentes, al menos, desde el inicio del brote de COVID-19 en ambos barrios respectivamente. Estos resultados representarían una circulación viral con poca o ninguna restricción intra-barrio.
-
En el resto de las muestras de los barrios de CABA también predominan los linajes B.1.3 y B.1.1.33, aunque el análisis filogenético mostró un agrupamiento circunstancial entre sí y con los barrios populares, lo que nuevamente indicaría una restricción en la transmisión viral entre los mismos.
Cabe destacar que en abril del corriente año, se secuenciaron nuevos genomas virales de SARS-CoV2 en 26 pacientes. En este trabajo se estudiaron virus provenientes de pacientes con historia de viaje (casos importados), virus provenientes de pacientes que tuvieron contacto directo con casos importados, y virus procedentes de pacientes que no tuvieron contacto confirmado con casos importados (circulación autóctona).
Asimismo, en un estudio realizado por laboratorios y centros del sur del país, científicos y científicas obtuvieron 57 nuevos genomas de SARS-CoV2 de pacientes de la Patagonia. Este reporte muestra el trabajo realizado en el nodo de secuenciación más austral del Consorcio, conformado por el Centro Austral de Investigaciones Científicas (CADIC-CONICET), el Hospital Regional de Ushuaia y la Universidad Nacional de Tierra del Fuego (UNTDF) ubicados en la ciudad de Ushuaia, provincia de Tierra del Fuego, Antártida e Islas del Atlántico Sur. El equipo de trabajo de este nodo está conformado por los Dres. Santiago Ceballos, IvanGramundi, Cristina Nardi y Fernando Gallego, con la colaboración del Dr. Daniel Fernández en el análisis de datos.
Como conclusión general se puede indicar que, si bien un mismo linaje viral puede encontrarse simultáneamente en distintas regiones geográficas, el patrón general observado es la introducción viral y diversificación zonal restringida, más que la circulación irrestricta de los linajes entre las distintas zonas.
En el próximo análisis del AMBA, que comprenderá los meses de junio y julio, podrán contrastarse estos resultados con los correspondientes a un paulatino aumento del movimiento de personas menos restringido entre zonas geográficas del AMBA, acompañado a un aumento progresivo del número de casos de la COVID-19.
Cabe destacar la potencialidad de los estudios filogenéticos y epidemiológicos conjuntos para el análisis tanto del patrón de circulación global como de cadenas de transmisión viral particulares, así como la importancia de mantener los estudios en el tiempo para dar continuidad al análisis del surgimiento, mantenimiento y desaparición de grupos filogenéticos y linajes virales.
Descargas
Consorcio (0.69 MB)
Descargar archivoReporte CABA (3.47 MB)
Descargar archivoReporte GBA (2.3 MB)
Descargar archivo