Administración Nacional de Medicamentos, Alimentos y Tecnología Médica
PRODUCTOS MEDICINALES
Disposición 1918/2013
Establécense los criterios para la selección de una especialidad medicinal como producto de referencia para los estudios de Bioequivalencia y Equivalencia In-Vitro.
Bs. As., 11/4/2013
VISTO la Ley Nº 16.463, sus Decretos Reglamentarios Nros. 9763/64, 150/92 (T.O. 1993), las Disposiciones (ANMAT) Nros. 6677/10, 3185/99, sus complementarias y modificatorias, 5040/06 (modificada por la Disposición ANMAT Nº 1746/07), 556/09 y 758/09 y el Expediente Nº 1-47-162-11-4 del registro de esta Administración Nacional de Medicamentos, Alimentos y Tecnología Médica y;
CONSIDERANDO:
Que el artículo 1° de la Ley 16.463 establece que “quedan sometidos a la presente ley y a los reglamentos que en su consecuencia se dicten, la importación, exportación, producción, elaboración, fraccionamiento, comercialización o depósito en jurisdicción nacional o con destino al comercio interprovincial de las drogas, productos químicos, reactivas, formas farmacéuticas, medicamentos, elementos de diagnóstico, y todo otro producto de uso y aplicación en medicina humana y las personas de existencia visible o ideal que intervengan en dichas actividades”.
Que el artículo 2° de la citada ley establece que las actividades mencionadas sólo podrán realizarse previa autorización y bajo el contralor de la autoridad sanitaria, en establecimientos por ella habilitados y bajo la dirección técnica del profesional universitario correspondiente, todo ello en las condiciones y dentro de las normas que establezca la reglamentación, atendiendo a las características particulares de cada actividad y a razonables garantías técnicas en salvaguarda de la salud pública y de la economía del consumidor.
Que asimismo el artículo 3° del mencionado cuerpo legal prescribe que los productos comprendidos en la citada ley deberán reunir las condiciones establecidas en la Farmacopea Argentina, y en caso de no figurar en ella, las que surgen de los patrones internacionales y de los textos de reconocido valor científico, debiendo a la vez ser inscriptos por ante esta Administración Nacional de conformidad a lo establecido en el Decreto Nº 150/92 (T.O. 1993).
Que el artículo 1° del Decreto Nº 9763/64, reglamentario de la Ley 16.463, establece que el ejercicio del poder de policía sanitaria referido a las actividades indicadas en el artículo 1° de la mentada ley, y a las personas de existencia visible o ideal que intervengan en las mismas, se hará efectivo por el Ministerio de Asistencia Social y Salud Pública de la Nación (hoy Ministerio de Salud), en las jurisdicciones que allí se indican.
Que por su parte el Decreto Nº 1490/92, crea esta Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT), como organismo descentralizado de la Administración Pública Nacional, con un régimen de autarquía financiera y económica, con jurisdicción en todo el territorio nacional, asumiendo dichas funciones.
Que en virtud del artículo 3°, inciso a) del mencionado decreto, esta Administración Nacional tiene competencia, entre otras materias, en todo lo referente al control y fiscalización sobre la sanidad y la calidad de las drogas, productos químicos, reactivos, formas farmacéuticas, medicamentos, elementos de diagnóstico, materiales y tecnologías biomédicas y todo otro producto de uso y aplicación en medicina humana.
Que esta Administración Nacional es la autoridad reguladora de medicamentos, y está facultada para otorgar su registro sanitario, de acuerdo a los requisitos y procedimientos establecidos en cada caso.
Que el Decreto Nº 150/92 (T.O. 1993), reglamentario de la Ley de Medicamentos Nº 16.463, estableció una serie de definiciones, normas y procedimientos, que constituyen la base sobre la cual se sustenta todo lo relacionado con el registro, elaboración, fraccionamiento, expendio, comercialización, exportación e importación de medicamentos y especialidades medicinales.
Que de conformidad a las prescripciones de dicho decreto se entiende por medicamento a “toda preparación o producto farmacéutico empleado para la prevención, diagnóstico y/o tratamiento de una enfermedad o estado patológico, o para modificar sistemas fisiológicos en beneficio de la persona a quien se le administra” (art. 1° inciso a).
Que por su parte el inciso b) del citado artículo define principio activo o droga farmacéutica como “toda sustancia o mezcla de sustancias relacionadas, de origen natural o sintético que poseyendo un efecto farmacológico específico, se emplea en medicina humana”.
Que asimismo el inciso c) define nombre genérico como “denominación de un principio activo o droga farmacéutica, o cuando corresponda, de una asociación o combinación de principios activos a dosis fijas, adoptada por la autoridad sanitaria nacional, o, en su defecto, la denominación común internacional de un principio activo recomendada por la Organización Mundial de la Salud”.
Que finalmente el inciso d) define especialidad medicinal como “todo medicamento designado por su nombre convencional, sea o no una marca de fábrica o comercial, o por el nombre genérico que corresponda a su composición y contenido, preparado y envasado uniformemente para su distribución y expendio, de composición cuantitativa definida, declarada y verificable, de forma farmacéutica estable y acción terapéutica comprobable”.
Que la aplicación de las normativas aludidas, y las mencionadas en el Visto de la presente disposición, tienen como finalidad última la protección de la salud de la población, mediante la adopción de un modelo fiscalizador de gestión, que sin perjuicio de la lectura objetiva de información calificada, destine los mayores esfuerzos a garantizar la eficacia, seguridad y calidad de los productos que aquélla consume.
Que mediante procesos de investigación y desarrollo se obtienen productos que se denominan originales o innovadores, incluyendo entre sus etapas la síntesis química, investigaciones preclínica, galénica y clínica, siendo esta última la que permite inferir su farmacocinética, biodisponibilidad, farmacodinámica, seguridad y eficacia terapéutica.
Que existen medicamentos que poseen los mismos ingredientes farmacéuticos activos (IFAs) que el medicamento innovador, pudiendo diferir en la composición de sus excipientes, y que cumplen con las Buenas Prácticas de Manufactura, almacenamiento y distribución, definiéndose a éstos como productos multifuente (OMS, Informe Técnico Nº 937, Anexo 7, del año 2006).
Que el fundamento científico de los estudios de bioequivalencia o de equivalencia in Vitro es la comparabilidad de sus biodisponiblidades o de sus perfiles de disolución, respectivamente, entre el producto multifuente y el producto de referencia.
Que por Disposición (ANMAT) Nº 3185/99, en su ANEXO I, punto II, “Definiciones”, se define al producto de referencia como aquel producto para el cual la eficacia y seguridad han sido establecidas.
Que por Disposiciones (ANMAT) Nros. 3185/99, 5040/06, y su modificatoria 1746/07 y 758/09 se han establecido los requerimientos técnicos y metodológicos para la realización de estudios de Bioequivalencia in-vivo y/o Equivalencia in Vitro entre una formulación multifuente y otra que esta ANMAT fija como de referencia.
Que a los fines de asegurar la continuidad de la calidad y del comportamiento de los productos designados por este organismo de control, como referencia para estudios de bioequivalencia, resulta necesario documentar sus características.
Que a fojas 1/3 obra el informe técnico de la Comisión Asesora “Ad Honorem” en temas de Bioequivalencia y Biodisponibilidad, creada por Disposición ANMAT Nº 4351/10.
Que la aludida Comisión Asesora, el Instituto Nacional de Medicamentos y la Dirección de Asuntos Jurídicos, han tomado la intervención de su competencia.
Que se actúa en virtud de las facultades conferidas por los Decretos Nros. 1490/92 y 425/10.
Por ello;
EL INTERVENTOR DE LA ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA
DISPONE:
Artículo 1° — Establécense los criterios para la selección de una especialidad medicinal como producto de referencia para los estudios de Bioequivalencia y Equivalencia In-Vitro que figuran en el Anexo I de la presente disposición, que forma parte integrante de la misma.
Art. 2° — Establécese que para la designación de una especialidad medicinal como producto de referencia para los estudios de Bioequivalencia y Equivalencia In-Vitro, ya sea solicitada por su titular o cuando así lo estime necesario esta Administración Nacional, deberá presentarse la documentación que obra en el Anexo II de la presente disposición, que forma parte integrante de la misma.
Art. 3° — Una vez aprobada la documentación mencionada en el artículo inmediato precedente, esta ANMAT designará el Producto de Referencia para estudios de Bioequivalencia y Equivalencia In-Vitro, que deban cumplimentar todas las formas farmacéuticas y las concentraciones de todos los productos similares / multifuentes comercializados a la fecha, incluyendo los establecidos en la Disp. ANMAT Nº 4788/12.
Art. 4° — Déjanse sin efecto los criterios de selección del producto de referencia establecidos en la Disposición (ANMAT) Nº 758/09, Anexo I, Punto IV.
Art. 5° — Apruébase el formulario índice para la presentación de la documentación necesaria para que una especialidad medicinal sea designada como producto de referencia (según lo establecido en el Anexo II), que obra en el Anexo III de la presente disposición y que forma parte integrante de la misma.
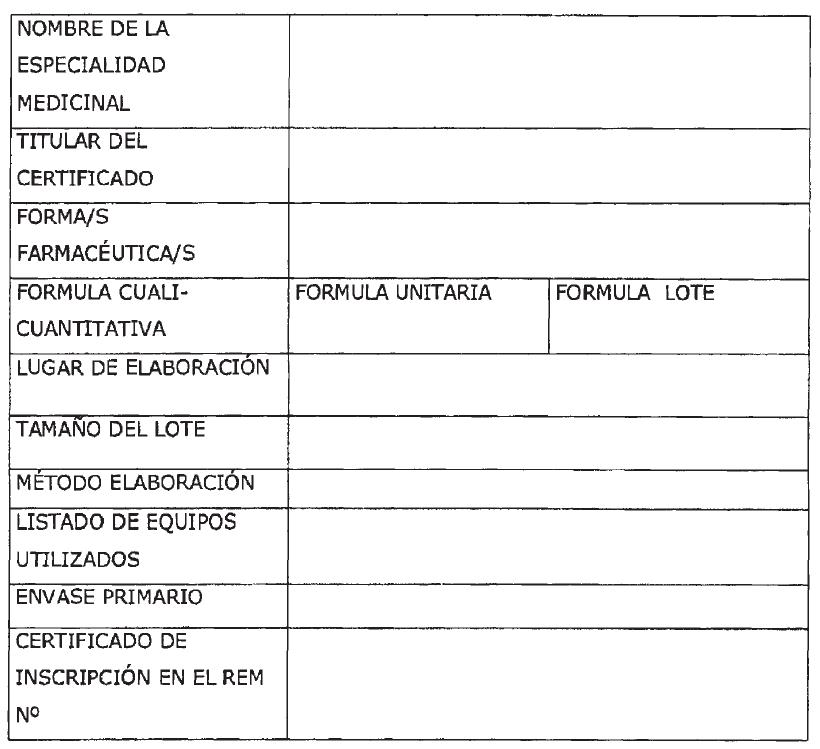
Art. 6° — Apruébase el formulario de caracterización del producto, que figura como Anexo IV de la presente disposición y que forma parte integrante de la misma.
Art. 7° — La presente Disposición entrará en vigencia a partir del primer día hábil siguiente al de su publicación en el Boletín Oficial.
Art. 8° — Regístrese. Dése a la Dirección Nacional del Registro Oficial para su publicación. Notifíquese a las Cámaras de Especialidades Medicinales (CILFA, CAEME, COOPERALA, CAPGEN, CAPEMVeL, SAFYBI), Confederación Médica de la República Argentina (COMRA) y la Confederación Farmacéutica Argentina (COFA). Cumplido archívese PERMANENTE. — Carlos Chiale.
Esta ANMAT seleccionará, según los criterios del presente ANEXO, para cada ingrediente farmacéutico activo, forma farmacéutica y concentración, al candidato a Producto de Referencia y notificará al laboratorio titular del Registro, el cual deberá presentar la documentación del Anexo II cuya aceptación determinará su designación como Producto de Referencia para estudios de Bioequivalencia y Equivalencia In-Vitro.
Se exceptúan de este procedimiento los productos comparadores del Item 2 del presente Anexo para los cuales esta ANMAT implementará las acciones necesarias para facilitar la disponibilidad del producto de Referencia.
Los criterios para la selección del Producto de Referencia son:
1° Producto Innovador consumido y comercializado en el país; o
2° Producto no consumido ni comercializado en el país, que:
2.1. Se encuentra incluido como producto comparador para estudios de bioequivalencia en el Informe Técnico de la OMS, Nº 902, del año 2002 y posteriores; o
2.2. Producto Innovador consumido y proveniente de un país ICH (International Conference on Harmonisation) o país asociado; o
3° Producto designado por esta ANMAT de acuerdo a su perfil de seguridad, eficacia y/o estudio/s de su comportamiento farmacocinético y toda otra Información que esta autoridad sanitaria estime correspondiente, ejemplo: antecedentes de farrnacovigilancla registrados a lo largo de su comercialización.
A continuación se presenta el algoritmo decisorio para establecer el producto de referencia.
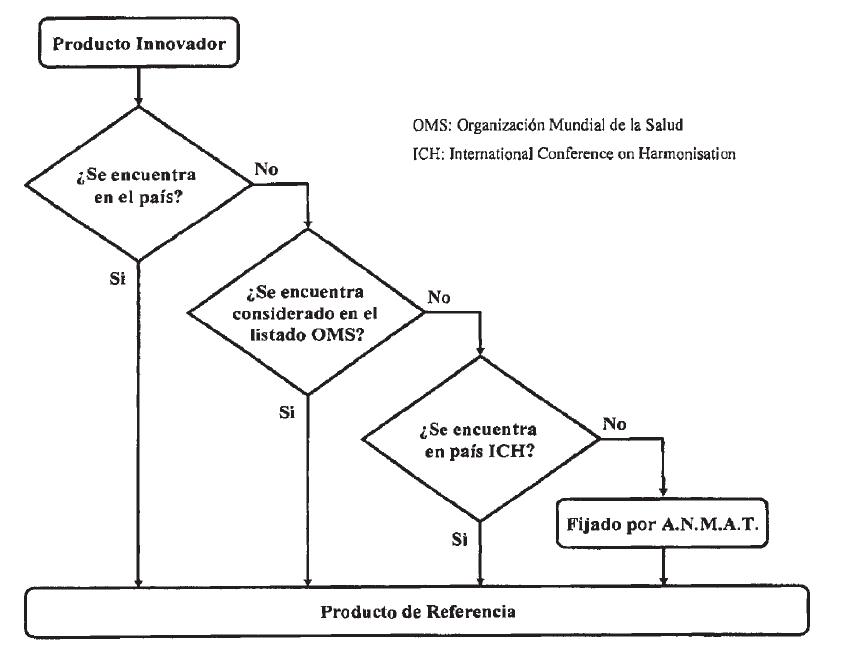
Arbol (algoritmo) de decisión para la designación de una especialidad medicinal como producto de referencia para estudios de bioequivalencia y/o equivalencia in-vitro.
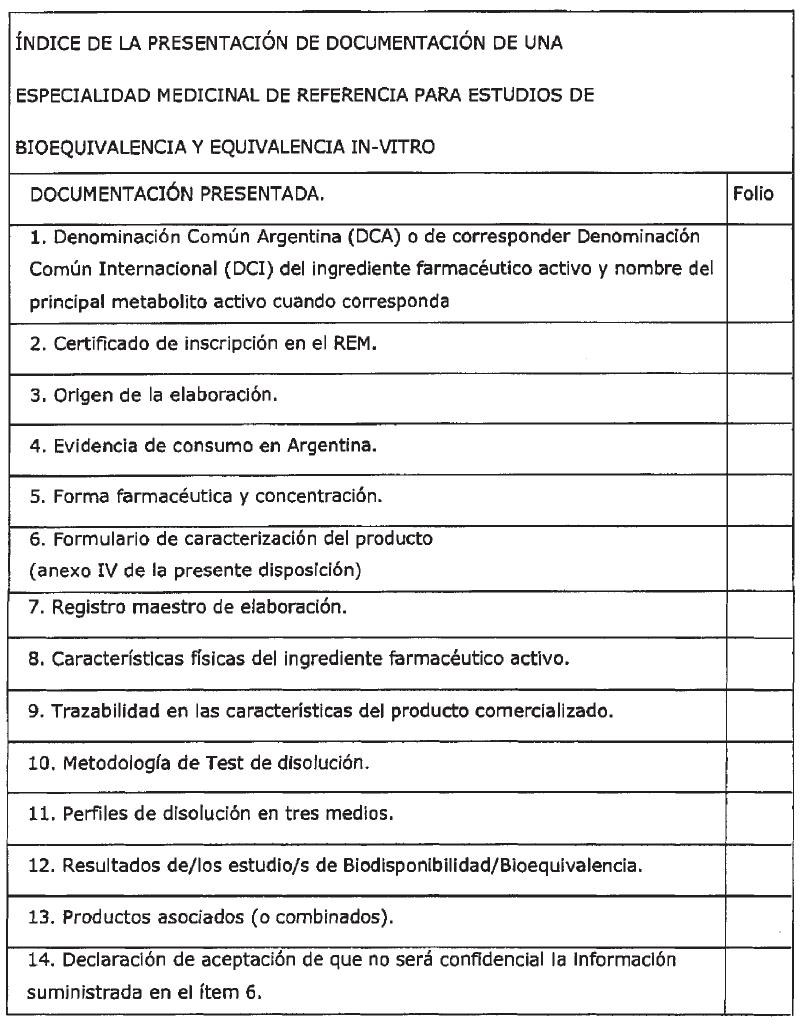
1. Denominación Común Argentina (DCA) o de corresponder Denominación Común Internacional (DCI) del ingrediente farmacéutico activo: incluir dicha denominación y la del principal metabolito activo cuando corresponda.
2. Certificado: copia del certificado de inscripción en el REM, incluidos cambios posteriores al registro.
3. Origen de la elaboración de cada etapa desde el IFA hasta el producto terminado: estableciendo claramente el país, empresa productora y dirección de la misma.
4. Evidencia de consumo en Argentina.
5. Formas farmacéuticas y concentraciones aprobadas.
6. Caracterización de fórmula: conforme al formulario que figura en el Anexo IV del presente.
7. Registro maestro de elaboración.
8. Características físicas del ingrediente farmacéutico activo:
8.1. forma cristalina.
8.2. solubilidad.
8.3. tamaño de partícula.
9. Trazabilidad en las condiciones de elaboración del producto actualmente comercializado, respecto al biolote.
10. Metodología del Test de disolución: coincidente con la aprobada y presentación de los resultados.
11. Perfiles de disolución de dos o más lotes, en tres medios: según lo establecido en la Disposición (ANMAT) Nº 758/09 con cálculo del factor de similaridad interlote.
12. Resultados del/los estudio/s de Biodisponibilidad/Bioequivalencia: correspondiente a la formulación seleccionada para consumir en Argentina, especificando el lote, con respecto al producto innovador que haya demostrado seguridad y eficacia inicialmente con el fin de demostrar que el producto a ser designado de referencia en el país está correlacionado de manera confiable con los datos clínicos originales, incluyendo:
12.1. presentación de los resultados según Disposición (ANMAT) Nº 5040/06 y su modificatoria 1746/09, ítem 4.9, Anexo I y Anexo IV.
12.2. cuando corresponda:
12.2.1. estudios realizados en ayunas y/o con alimentos.
12.2.2. principal metabolito activo.
12.2.3. estudios puente entre fase II - III y la formulación comercializada.
12.2.4. estudios para cada forma farmacéutica y concentración que lo requiera.
13. Productos asociados (o combinados). Incluir el perfil farmacocinético de los fármacos:
13.1. En forma individual.
13.2. De la asociación.
14. Declaración de aceptación de que no será confidencial la información suministrada en el ítem 6.
PRODUCTOS MEDICINALES
Disposición 1918/2013
Establécense los criterios para la selección de una especialidad medicinal como producto de referencia para los estudios de Bioequivalencia y Equivalencia In-Vitro.
Bs. As., 11/4/2013
VISTO la Ley Nº 16.463, sus Decretos Reglamentarios Nros. 9763/64, 150/92 (T.O. 1993), las Disposiciones (ANMAT) Nros. 6677/10, 3185/99, sus complementarias y modificatorias, 5040/06 (modificada por la Disposición ANMAT Nº 1746/07), 556/09 y 758/09 y el Expediente Nº 1-47-162-11-4 del registro de esta Administración Nacional de Medicamentos, Alimentos y Tecnología Médica y;
CONSIDERANDO:
Que el artículo 1° de la Ley 16.463 establece que “quedan sometidos a la presente ley y a los reglamentos que en su consecuencia se dicten, la importación, exportación, producción, elaboración, fraccionamiento, comercialización o depósito en jurisdicción nacional o con destino al comercio interprovincial de las drogas, productos químicos, reactivas, formas farmacéuticas, medicamentos, elementos de diagnóstico, y todo otro producto de uso y aplicación en medicina humana y las personas de existencia visible o ideal que intervengan en dichas actividades”.
Que el artículo 2° de la citada ley establece que las actividades mencionadas sólo podrán realizarse previa autorización y bajo el contralor de la autoridad sanitaria, en establecimientos por ella habilitados y bajo la dirección técnica del profesional universitario correspondiente, todo ello en las condiciones y dentro de las normas que establezca la reglamentación, atendiendo a las características particulares de cada actividad y a razonables garantías técnicas en salvaguarda de la salud pública y de la economía del consumidor.
Que asimismo el artículo 3° del mencionado cuerpo legal prescribe que los productos comprendidos en la citada ley deberán reunir las condiciones establecidas en la Farmacopea Argentina, y en caso de no figurar en ella, las que surgen de los patrones internacionales y de los textos de reconocido valor científico, debiendo a la vez ser inscriptos por ante esta Administración Nacional de conformidad a lo establecido en el Decreto Nº 150/92 (T.O. 1993).
Que el artículo 1° del Decreto Nº 9763/64, reglamentario de la Ley 16.463, establece que el ejercicio del poder de policía sanitaria referido a las actividades indicadas en el artículo 1° de la mentada ley, y a las personas de existencia visible o ideal que intervengan en las mismas, se hará efectivo por el Ministerio de Asistencia Social y Salud Pública de la Nación (hoy Ministerio de Salud), en las jurisdicciones que allí se indican.
Que por su parte el Decreto Nº 1490/92, crea esta Administración Nacional de Medicamentos, Alimentos y Tecnología Médica (ANMAT), como organismo descentralizado de la Administración Pública Nacional, con un régimen de autarquía financiera y económica, con jurisdicción en todo el territorio nacional, asumiendo dichas funciones.
Que en virtud del artículo 3°, inciso a) del mencionado decreto, esta Administración Nacional tiene competencia, entre otras materias, en todo lo referente al control y fiscalización sobre la sanidad y la calidad de las drogas, productos químicos, reactivos, formas farmacéuticas, medicamentos, elementos de diagnóstico, materiales y tecnologías biomédicas y todo otro producto de uso y aplicación en medicina humana.
Que esta Administración Nacional es la autoridad reguladora de medicamentos, y está facultada para otorgar su registro sanitario, de acuerdo a los requisitos y procedimientos establecidos en cada caso.
Que el Decreto Nº 150/92 (T.O. 1993), reglamentario de la Ley de Medicamentos Nº 16.463, estableció una serie de definiciones, normas y procedimientos, que constituyen la base sobre la cual se sustenta todo lo relacionado con el registro, elaboración, fraccionamiento, expendio, comercialización, exportación e importación de medicamentos y especialidades medicinales.
Que de conformidad a las prescripciones de dicho decreto se entiende por medicamento a “toda preparación o producto farmacéutico empleado para la prevención, diagnóstico y/o tratamiento de una enfermedad o estado patológico, o para modificar sistemas fisiológicos en beneficio de la persona a quien se le administra” (art. 1° inciso a).
Que por su parte el inciso b) del citado artículo define principio activo o droga farmacéutica como “toda sustancia o mezcla de sustancias relacionadas, de origen natural o sintético que poseyendo un efecto farmacológico específico, se emplea en medicina humana”.
Que asimismo el inciso c) define nombre genérico como “denominación de un principio activo o droga farmacéutica, o cuando corresponda, de una asociación o combinación de principios activos a dosis fijas, adoptada por la autoridad sanitaria nacional, o, en su defecto, la denominación común internacional de un principio activo recomendada por la Organización Mundial de la Salud”.
Que finalmente el inciso d) define especialidad medicinal como “todo medicamento designado por su nombre convencional, sea o no una marca de fábrica o comercial, o por el nombre genérico que corresponda a su composición y contenido, preparado y envasado uniformemente para su distribución y expendio, de composición cuantitativa definida, declarada y verificable, de forma farmacéutica estable y acción terapéutica comprobable”.
Que la aplicación de las normativas aludidas, y las mencionadas en el Visto de la presente disposición, tienen como finalidad última la protección de la salud de la población, mediante la adopción de un modelo fiscalizador de gestión, que sin perjuicio de la lectura objetiva de información calificada, destine los mayores esfuerzos a garantizar la eficacia, seguridad y calidad de los productos que aquélla consume.
Que mediante procesos de investigación y desarrollo se obtienen productos que se denominan originales o innovadores, incluyendo entre sus etapas la síntesis química, investigaciones preclínica, galénica y clínica, siendo esta última la que permite inferir su farmacocinética, biodisponibilidad, farmacodinámica, seguridad y eficacia terapéutica.
Que existen medicamentos que poseen los mismos ingredientes farmacéuticos activos (IFAs) que el medicamento innovador, pudiendo diferir en la composición de sus excipientes, y que cumplen con las Buenas Prácticas de Manufactura, almacenamiento y distribución, definiéndose a éstos como productos multifuente (OMS, Informe Técnico Nº 937, Anexo 7, del año 2006).
Que el fundamento científico de los estudios de bioequivalencia o de equivalencia in Vitro es la comparabilidad de sus biodisponiblidades o de sus perfiles de disolución, respectivamente, entre el producto multifuente y el producto de referencia.
Que por Disposición (ANMAT) Nº 3185/99, en su ANEXO I, punto II, “Definiciones”, se define al producto de referencia como aquel producto para el cual la eficacia y seguridad han sido establecidas.
Que por Disposiciones (ANMAT) Nros. 3185/99, 5040/06, y su modificatoria 1746/07 y 758/09 se han establecido los requerimientos técnicos y metodológicos para la realización de estudios de Bioequivalencia in-vivo y/o Equivalencia in Vitro entre una formulación multifuente y otra que esta ANMAT fija como de referencia.
Que a los fines de asegurar la continuidad de la calidad y del comportamiento de los productos designados por este organismo de control, como referencia para estudios de bioequivalencia, resulta necesario documentar sus características.
Que a fojas 1/3 obra el informe técnico de la Comisión Asesora “Ad Honorem” en temas de Bioequivalencia y Biodisponibilidad, creada por Disposición ANMAT Nº 4351/10.
Que la aludida Comisión Asesora, el Instituto Nacional de Medicamentos y la Dirección de Asuntos Jurídicos, han tomado la intervención de su competencia.
Que se actúa en virtud de las facultades conferidas por los Decretos Nros. 1490/92 y 425/10.
Por ello;
EL INTERVENTOR DE LA ADMINISTRACION NACIONAL DE MEDICAMENTOS, ALIMENTOS Y TECNOLOGIA MEDICA
DISPONE:
Artículo 1° — Establécense los criterios para la selección de una especialidad medicinal como producto de referencia para los estudios de Bioequivalencia y Equivalencia In-Vitro que figuran en el Anexo I de la presente disposición, que forma parte integrante de la misma.
Art. 2° — Establécese que para la designación de una especialidad medicinal como producto de referencia para los estudios de Bioequivalencia y Equivalencia In-Vitro, ya sea solicitada por su titular o cuando así lo estime necesario esta Administración Nacional, deberá presentarse la documentación que obra en el Anexo II de la presente disposición, que forma parte integrante de la misma.
Art. 3° — Una vez aprobada la documentación mencionada en el artículo inmediato precedente, esta ANMAT designará el Producto de Referencia para estudios de Bioequivalencia y Equivalencia In-Vitro, que deban cumplimentar todas las formas farmacéuticas y las concentraciones de todos los productos similares / multifuentes comercializados a la fecha, incluyendo los establecidos en la Disp. ANMAT Nº 4788/12.
Art. 4° — Déjanse sin efecto los criterios de selección del producto de referencia establecidos en la Disposición (ANMAT) Nº 758/09, Anexo I, Punto IV.
Art. 5° — Apruébase el formulario índice para la presentación de la documentación necesaria para que una especialidad medicinal sea designada como producto de referencia (según lo establecido en el Anexo II), que obra en el Anexo III de la presente disposición y que forma parte integrante de la misma.
Art. 6° — Apruébase el formulario de caracterización del producto, que figura como Anexo IV de la presente disposición y que forma parte integrante de la misma.
Art. 7° — La presente Disposición entrará en vigencia a partir del primer día hábil siguiente al de su publicación en el Boletín Oficial.
Art. 8° — Regístrese. Dése a la Dirección Nacional del Registro Oficial para su publicación. Notifíquese a las Cámaras de Especialidades Medicinales (CILFA, CAEME, COOPERALA, CAPGEN, CAPEMVeL, SAFYBI), Confederación Médica de la República Argentina (COMRA) y la Confederación Farmacéutica Argentina (COFA). Cumplido archívese PERMANENTE. — Carlos Chiale.
ANEXO I
CRITERIOS DE SELECCION PARA LA DESIGNACION DE UNA ESPECIALIDAD MEDICINAL COMO PRODUCTO DE REFERENCIA PARA ESTUDIOS DE BIOEQUIVALENCIA Y EQUIVALENCIA IN-VITRO
Esta ANMAT seleccionará, según los criterios del presente ANEXO, para cada ingrediente farmacéutico activo, forma farmacéutica y concentración, al candidato a Producto de Referencia y notificará al laboratorio titular del Registro, el cual deberá presentar la documentación del Anexo II cuya aceptación determinará su designación como Producto de Referencia para estudios de Bioequivalencia y Equivalencia In-Vitro.
Se exceptúan de este procedimiento los productos comparadores del Item 2 del presente Anexo para los cuales esta ANMAT implementará las acciones necesarias para facilitar la disponibilidad del producto de Referencia.
Los criterios para la selección del Producto de Referencia son:
1° Producto Innovador consumido y comercializado en el país; o
2° Producto no consumido ni comercializado en el país, que:
2.1. Se encuentra incluido como producto comparador para estudios de bioequivalencia en el Informe Técnico de la OMS, Nº 902, del año 2002 y posteriores; o
2.2. Producto Innovador consumido y proveniente de un país ICH (International Conference on Harmonisation) o país asociado; o
3° Producto designado por esta ANMAT de acuerdo a su perfil de seguridad, eficacia y/o estudio/s de su comportamiento farmacocinético y toda otra Información que esta autoridad sanitaria estime correspondiente, ejemplo: antecedentes de farrnacovigilancla registrados a lo largo de su comercialización.
A continuación se presenta el algoritmo decisorio para establecer el producto de referencia.
Arbol (algoritmo) de decisión para la designación de una especialidad medicinal como producto de referencia para estudios de bioequivalencia y/o equivalencia in-vitro.
ANEXO II
DOCUMENTACION QUE DEBE PRESENTAR EL LABORATORIO TITULAR DE UNA ESPECIALIDAD MEDICINAL PARA COMPLETAR EL FORMULARIO INDICE (ANEXO III) PARA SU EVALUACION COMO PRODUCTO DE REFERENCIA
1. Denominación Común Argentina (DCA) o de corresponder Denominación Común Internacional (DCI) del ingrediente farmacéutico activo: incluir dicha denominación y la del principal metabolito activo cuando corresponda.
2. Certificado: copia del certificado de inscripción en el REM, incluidos cambios posteriores al registro.
3. Origen de la elaboración de cada etapa desde el IFA hasta el producto terminado: estableciendo claramente el país, empresa productora y dirección de la misma.
4. Evidencia de consumo en Argentina.
5. Formas farmacéuticas y concentraciones aprobadas.
6. Caracterización de fórmula: conforme al formulario que figura en el Anexo IV del presente.
7. Registro maestro de elaboración.
8. Características físicas del ingrediente farmacéutico activo:
8.1. forma cristalina.
8.2. solubilidad.
8.3. tamaño de partícula.
9. Trazabilidad en las condiciones de elaboración del producto actualmente comercializado, respecto al biolote.
10. Metodología del Test de disolución: coincidente con la aprobada y presentación de los resultados.
11. Perfiles de disolución de dos o más lotes, en tres medios: según lo establecido en la Disposición (ANMAT) Nº 758/09 con cálculo del factor de similaridad interlote.
12. Resultados del/los estudio/s de Biodisponibilidad/Bioequivalencia: correspondiente a la formulación seleccionada para consumir en Argentina, especificando el lote, con respecto al producto innovador que haya demostrado seguridad y eficacia inicialmente con el fin de demostrar que el producto a ser designado de referencia en el país está correlacionado de manera confiable con los datos clínicos originales, incluyendo:
12.1. presentación de los resultados según Disposición (ANMAT) Nº 5040/06 y su modificatoria 1746/09, ítem 4.9, Anexo I y Anexo IV.
12.2. cuando corresponda:
12.2.1. estudios realizados en ayunas y/o con alimentos.
12.2.2. principal metabolito activo.
12.2.3. estudios puente entre fase II - III y la formulación comercializada.
12.2.4. estudios para cada forma farmacéutica y concentración que lo requiera.
13. Productos asociados (o combinados). Incluir el perfil farmacocinético de los fármacos:
13.1. En forma individual.
13.2. De la asociación.
14. Declaración de aceptación de que no será confidencial la información suministrada en el ítem 6.
ANEXO III
FORMULARIO INDICE DE LA PRESENTACION DE DOCUMENTACION POR EL LABORATORIO TITULAR DE UNA ESPECIALIDAD MEDICINAL PARA SU EVALUACION / DESIGNACION COMO PRODUCTO DE REFERENCIA PARA ESTUDIOS DE BIOEQUIVALENCIA Y/O EQUIVALENCIA IN-VITRO
ANEXO IV
FORMULARIO DE CARACTERIZACION DEL PRODUCTO